forum@abinit.org
Subject: The ABINIT Users Mailing List ( CLOSED )
List archive
- From: "Corsin Battaglia" <corsin.battaglia@freesurf.ch>
- To: <forum@abinit.org>
- Subject: fuzzy bandstructure
- Date: Sun, 1 Feb 2004 21:30:02 +0100
|
Dear abinit users
I am trying to calculate the bandstructure for the
metallic compound NbS2.
In a first step, I computed the total
energy. I used the Hartwigsen-Goedecker-Hutter pseudopotentials for Nb and
S. For Nb, I used the one, which contains semicore states as well (I read
somewhere that semicore states are necessary in an electronegative
environment).
During the scf-cycle, there seem to be no
convergence problems.
iter Etot(hartree) deltaE(h)
residm vres2 diffor
maxfor
ETOT 14 -154.22694726148 -3.875E-11 1.106E-04 8.090E-09 1.155E-06 2.924E-02 ETOT 15 -154.22694726148 -2.394E-12 1.110E-04 8.903E-10 2.442E-07 2.925E-02 ETOT 16 -154.22694726148 -9.612E-13 4.247E-05 5.884E-10 3.020E-07 2.925E-02 ETOT 17 -154.22694726148 -4.440E-13 3.888E-05 6.675E-12 1.490E-07 2.925E-02 The Fermi energy converges as
well
11792: newocc : new Fermi energy is 0.247380 , with nelect= 50.000000 12728: newocc : new Fermi energy is 0.247382 , with nelect= 50.000000 13664: newocc : new Fermi energy is 0.247381 , with nelect= 50.000000 14600: newocc : new Fermi energy is 0.247381 , with nelect= 50.000000 15536: newocc : new Fermi energy is 0.247381 , with nelect= 50.000000 16472: newocc : new Fermi energy is 0.247381 , with nelect= 50.000000 17408: newocc : new Fermi energy is 0.247381 , with nelect= 50.000000 No warnings or errors up to this point.
In a second step, I calculated the bandstructure. When I plot the
bandstructure, it looks very fuzzy, although the number of k-points was quite
dense (50 k-points between Gamma and M).
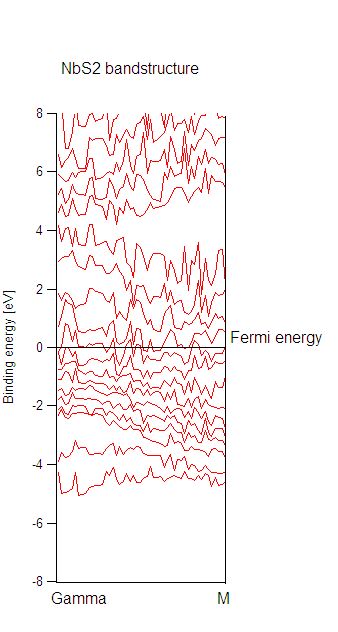 For every k-point, abinit writes the following warning into the log
file
17759: vtowfk: WARNING -
17760- Wavefunctions not converged for nnsclo,ikpt= 1 1 max resid= 1.22992E+00 Another problem is that abinit stops after 50 k-points, although I intended
to compute 125 k-points.
I have no clue, what I am doing wrong.
Thanks in advance for your help
Corsin
Here's are the files I used.
NbS2.files
NbS2.in
NbS2.out NbS2i NbS2o NbS2 ../../Pseudopotentials/41nb.13.hgh ../../Pseudopotentials/16s.6.hgh NbS2.in # 2H-NbS2 : computation of the total energy and
bandstructure
#Definition of the unit cell
acell 3.31 3.31 11.89 angstrom #rprim 0.866 -0.500 0.000 # It is better to define # 0.000 1.000 0.000 # the primitive vectors # 0.000 0.000 1.000 # using rprim angdeg 90 90 120 #Definition of the atom types
ntypat 2 # There are two type of atoms znucl 41 16 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. #Definition of the atoms natom 6 # There are six atoms natrd 2 # Reads two atoms typat 1 2 # type 1 is Nb, type 2 is S xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 1/4 # Triplet giving the REDUCED coordinate of atom 1. 1/3 2/3 1/8 # Triplet giving the REDUCED coordinate of atom 2. # Note the use of fractions (remember the limited # interpreter capabilities of ABINIT) spgroup 194 #Definition of the occupation numbers
occopt 4 tsmear 0.01 #Read psp
npsp 2 # Read 2 psp files ixc 1 # Nb is of type ixc 1. S is of type ixc 1. # LDA. Nb contains semicores. #Definition of the planewave basis set ecut 10.0 # Maximal kinetic energy cut-off, in Hartree ndtset 2
#Dataset 1: SCF GS calculation
#Definition of the k-point grid
kptopt1 1 # Option for the automatic generation of k points, taking # into account the symmetry ngkpt1 8 8 4 # This is the grid based on the primitive vectors # of the reciprocal space #Definition of the SCF procedure iscf1 3 # SCF cycle, CG based on the minim of the energy nstep1 250 # Maximal number of SCF cycles toldfe1 1.0d-12 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) #diemac 12.0 # For metals, we use the default 10^6. nband1 35 # nband=nb of electrons in unit cell/2+(20% for metals) # more bands are needed with semicore states prtden1 1 # Print the density for use by dataset 2 #Dataset 2: band structure
iscf2 -2 # Non-SCF
calculation
getden2 -1 kptopt2 -4 # Bandstructure with 4 lines nband2 35 # 35 bands ndivk2 50 25 50 10 kptbounds2 0.0 0.0 0.0 # Gamma point 0.5 0.0 0.0 # M point 2/3 2/3 0.0 # K point 0.0 0.0 0.0 # Gamma point 0.0 0.0 0.5 # A point tolwfr2 1.0d-12 # Only admitted convergence criterion for non-SCF calculations enunit2 1 # Output the eigenvalues in eV |
- fuzzy bandstructure, Corsin Battaglia, 02/01/2004
- Re: [abinit-forum] fuzzy bandstructure, verstraete, 02/02/2004
Archive powered by MHonArc 2.6.16.